IN A NUTSHELL
About This Work
Fungal pathogens cause fatal plant diseases, contributing to up to 20% loss in annual crop yield. These pathogens accomplish this using an arsenal of secreted effectors that can target the plant apoplast or can be translocated into the plant to affect cellular functions and disrupt host defences. Identification of fungal effectors is a crucial step, not only to advance our understanding of plant–pathogen interactions, but also to assist in developing novel disease prevention strategies to ultimately mitigate crop losses and improve food safety. However, the fast evolving nature of fungal effectors, coupled with the limited number of known fungal effectors, complicates efforts to reliably distinguish them from non-effectors.
Here, we introduce Fungtion (Fungal effector prediction), a toolkit leveraging a hybrid framework to accurately predict and visualize fungal effectors. By combining global patterns learned from pretrained protein language models with refined information from known effectors, Fungtion achieves state-of-the-art prediction performance. Additionally, the interactive visualizations we have developed enable researchers to explore both sequence- and high-level relationships between the predicted and known effectors, facilitating effector function discovery, annotation, and hypothesis formulation regarding plant-pathogen interactions. We anticipate Fungtion to be a valuable resource for biologists seeking deeper insights into fungal effector functions and for computational biologists aiming to develop future methodologies for fungal effector prediction.
ARCHITECTURE
The overall workflow of Fungtion is presented as follows: how we collected and curated the datasets, encoded features, trained models, evaluated model performance, and developed the toolkit.
1. Dataset
We gathered a total of 394 experimentally validated fungal effectors for our initial positive training dataset base on the positive training sets of previous prediction tools, including EffHunter, EffectorP 3.0, and Effector-GAN. The original independent positive set, totalling 100 entries, comprised independent positive sets from EffHunter, EffectorP 2.0 & 3.0, and Effector-GAN, as well as 22 novel fungal effectors manually curated from the literature. The original negative training set (7458 entries) and independent test set (3777 entries) were sourced from the negative training set and negative test set of Effector-GAN, respectively. After reducing sequence similarity within these datasets (using CD-HIT at a threshold of 40%) and structure similarity of positive samples between training and independent datasets using ECOD, we obtained a final training dataset comprising 178 positive and 6758 negative sequences, and an independent test set consisting of 34 positive and 3070 negative sequences. Additionally, three recently discovered novel, and hence independent, fungal effectors identified from the literature were used as case studies to examine the predicting capability of the proposed method.
2. Feature Encoding
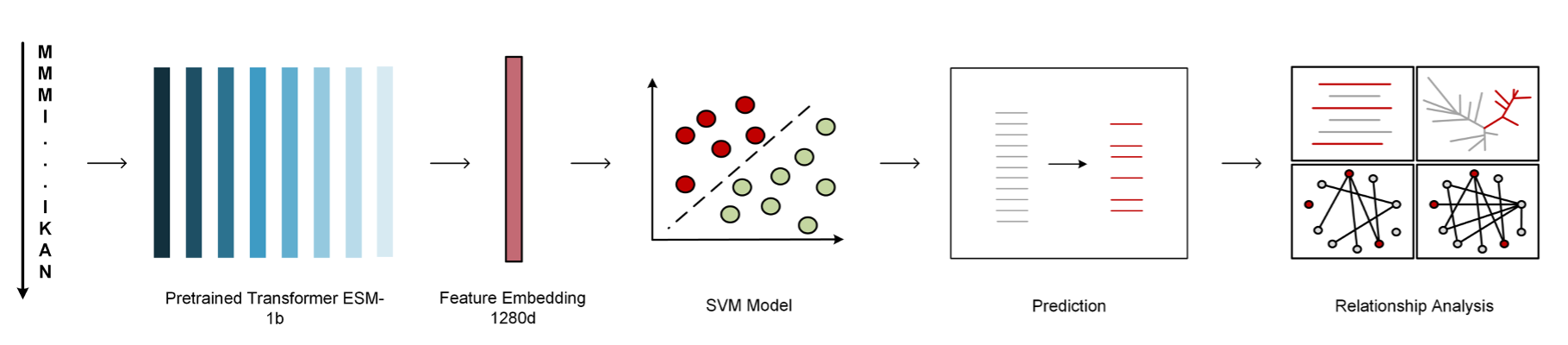
Instead of extracting features directly from the protein sequences, pretrained protein language models learn global patterns from a universally vast resource of proteins. Here, we utilized one pretrained protein language model ( ESM-1b ) to generate pretrain features.
3. Model Construction
Our imbalanced training dataset reflects the reality where the number of fungal effector proteins is usually much smaller than that of non-effector proteins in a fungal genome. To tackle the imbalanced between the positive and negative sequences, we created five subsets, each combining positive sequences with an equal number of randomly selected negative sequences from the training datasets. For each feature, we trained a classifier on each of the five balanced datasets using the classical Support Vector Machine (SVM) and constructed an ensemble model for the feature by averaging the prediction scores of the five classifiers. The Gaussian radial basis function (RBF) kernel was used in the SVM models to handle nonlinear biological sequence classification, and we fine-tuned the parameters Cost and Gamma through a grid search within the space {2-10,...,210} to optimize the SVM-based classifiers.
4. Performance Evaluation
The Fungtion predictor was extensively validated, with the baseline models and existing state-of-the-art tools on the 5-fold cross-validation and independent tests. Five performance metrics were used, including Sensitivity (SN), Specificity (SP), Accuracy (ACC), F-value and Matthews correlation coefficient (MCC). For each model, 5-fold cross-validation tests were conducted 5 times based on 5 balanced training datasets, and then the performance metrics were averaged as the final performance on the 5-fold cross-validation test.
5. Server Construction and usage
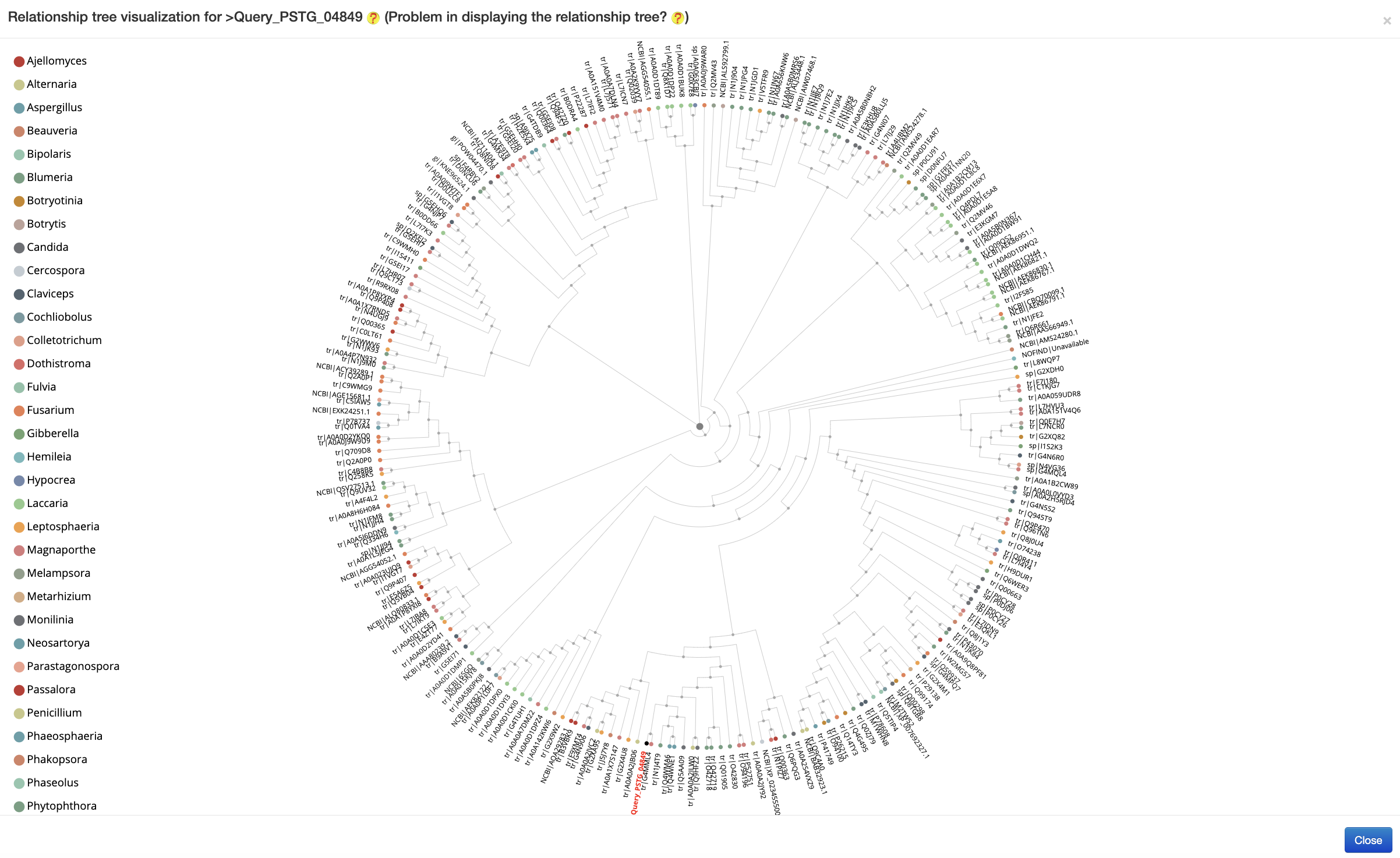
Like our previous servers ( AcrHub or PaCRISPR ), Fungtion consists of a prediction module to predict new fungal effectors and four relationship analysis modules to explore their connections to experimentally validated effectors. In the prediction module, Fungtion uses the ESM-1b model to identify potential fungal effectors from protein sequences. The predictor checks submitted sequences against experimentally validated fungal effectors, marking identical sequences with ‘Exp.’ and providing external links to detailed information in UniProtKB, while the remaining sequences undergo prediction. Among the four analysis modules, the similarity analysis module calculates sequence similarities between the predicted effector sequence and experimentally validated ones, displaying homologs with alignment scores and essential details, including query coverage, E-value, and Identity. Clicking on each matching effector sequence in the result table, it will reveal below the table the pairwise sequence alignment between the query protein and the corresponding experimentally validated fungal effector protein. Two similarity network analysis modules identify close sequence- and high-level relationships, respectively, within interactive networks that categorize nodes by species. The predicted effector is highlighted, and links between nodes indicate relationships. In the homology networks, users can hover for summaries or click for details in UniProtKB. Lastly, the relationship tree analysis module determines the closest relationships of the predicted effector with validated ones through an interactive relationship tree. Validated effectors are categorized by species, with the predicted sequence highlighted in red for easy identification. Hovering over proteins provides summaries, while clicking directs users to detailed descriptions in UniProtKB.
ONLINE WEB SERVER
1. Fungtion
To maximize user convenience, particularly for the amenability of biomedical scientists and biotechnologists, this user-friendly and publicly accessible web server has been established for the wider research community to perform predictions of novel putative fungal effectors from fungal genome-scale sequences (in format of protein sequences). Fungtion is a universal toolkit for predicting fungal effectors, with an extended visualization to infer their possible subtypes based on the sequence similarity, similarity network analysis modules and relationship tree analysis, freely accessible at https://step3.erc.monash.edu/Fungtion/.
1.1 Important settings of the Fungtion server
- The ensemble model currently used in the Fungtion server is trained based on the training dataset in line with its paper. It will be updated by combining all the experimentally fungal effectors as the positive dataset to retrain the prediction model. This operation would be continuously conducted through the retrieval of new experimentally validated fungal effectors on a regular basis.
2. Using the Fungtion web server
Navigate to the Fungtion prediction page by selecting 'Server' in the navigation menu or 'Go to use it! >' at the home page.
2.1 Input Formats
Two types of input are accepted by Fungtion: sequences in FASTA format (strongly recommended) and raw sequences. You can upload a file in FASTA format with your sequences, or insert them manually into the text box. For this tutorial, we will use an example list of 6 proteins. Click the example button and then submit.
In the case of input sequences in the FASTA format, you can prepare and input them as follows:
In addition, the following input sequence, which is in the original format downloadable from the UniProt database:
EGDLDFLKSHSKIIKTYAVSDCNTLQNLGPAAEAEGFQIQLGIWPNDDAHFEAEKEALQNYLPKISVSTIKIFLVGSEALYREDLTASELASKINDIKGLVKGIKGKNGKSYSSVPVGTVDSWDVLVDGASKPAIDAADVVYSNSFSYWQKNSQANASYSLFDDVMQALQTLQTAKGSTDIEFWVGETGWPTDGSSYGDSVPSVENAADQWQKGICALRAWGINVAVYEAFDEAWKPDTSGTSSVEKHWGVWQSDKTLKYSIDCKFN>P52751 MFSLKTVVLALAAAAFVQAIPAPGEGPSVSMAQQKCGAEKVVSCCNSKELKNSKSGAEIPIDVLSGECKNIPINILTINQLIPINNFCSDTVSCCSGEQIGLVNIQCTPILS>CSEP-07 virulenceMKLMQLIFYMLNILALTMAHNCCELSKMPTLPEIGVMSS
KPVRPKPIQSTASKGTKFLPTTLCIIKCKFNICLAHANVINSVQIGNRMRESKGAVNLNAPRHHSF
will be formatted (in order to remove those line breaks within the sequence) as follows:
In the case of raw sequences, you can input them as follows:
which will be formated by Fungtion as follows:
2.2 Input sequence limits
- Each sequence should have between 30 and 5000 amino acids.
- You can input up to 5000 sequences at once.
3. Fungtion Prediction Result Instructions
Fungtion contains a built-in list (continuously updated to keep in pace with new experimentally validated fungal effectors) of fungal effectors, as to annotate the prediction results after jobs are processed, through which we aim to distinguish the known fungal effectors from the computationally predicted ones.
For known fungal effectors, the results are marked as Exp. (an example is provided in the following figure).
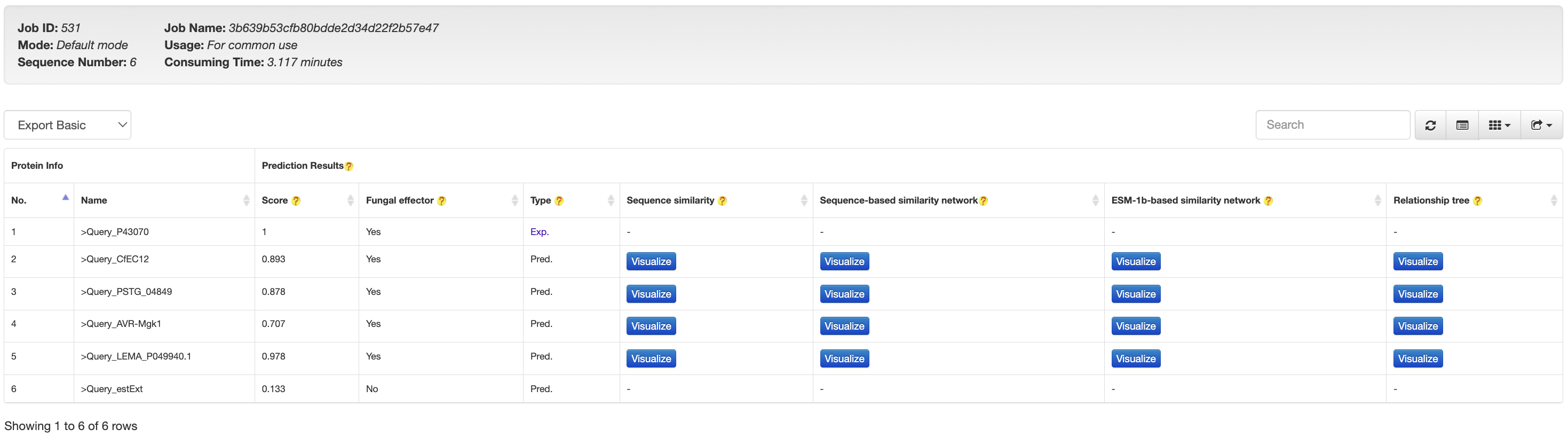
For a computationally predicted protein (fungal effector or non-fungal effector), the results are marked as Pred., while the detailed prediction results (predicted by our final model) will also be presented to users.To ease downstream analysis, each predicted fungal effector will be visualized in the format of sequence similarity, two similarity networks and relationship tree analysis with all known fungal effectors for users to infer its possible subtypes or functions.
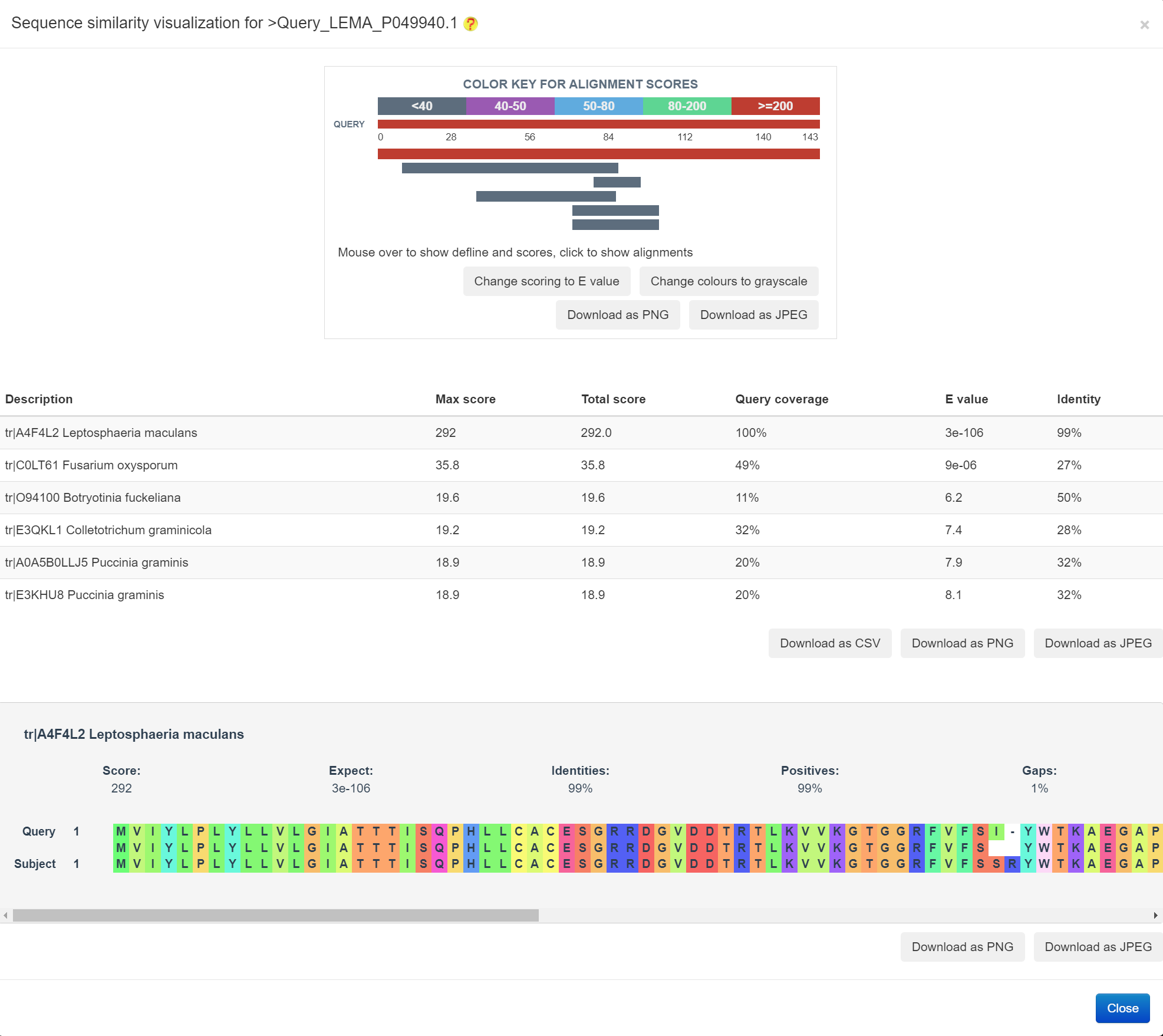
The sequence similarity relationship analysis was conducted by the blast-2.8.1+ program, followed by the visualization using the BLASTERJS library (an example is provided in the following figure)
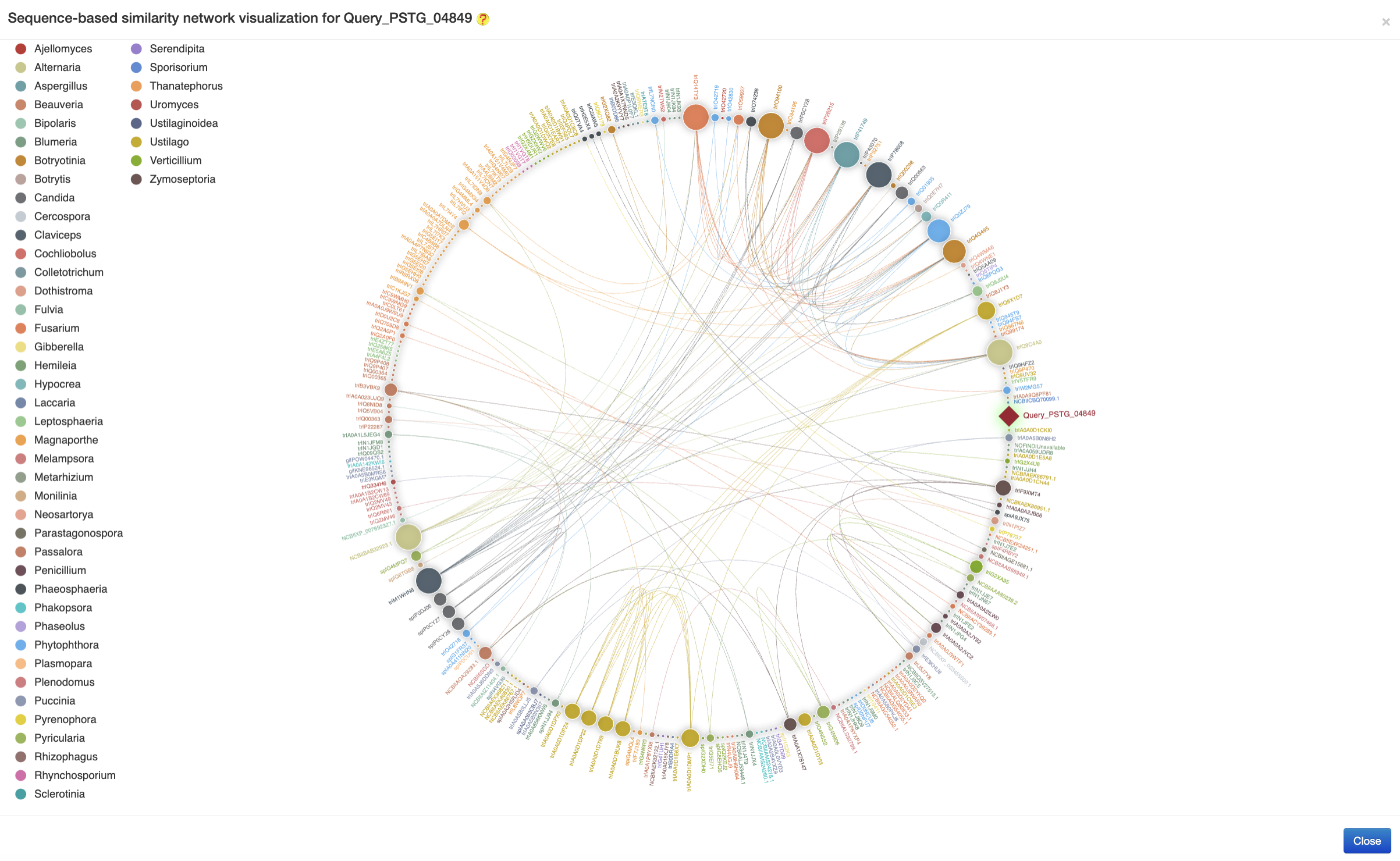
The sequence similarity network relationship analysis was conducted by the blast-2.2.26 program, followed by the visualization using the ECharts (an example is provided in the following figure)
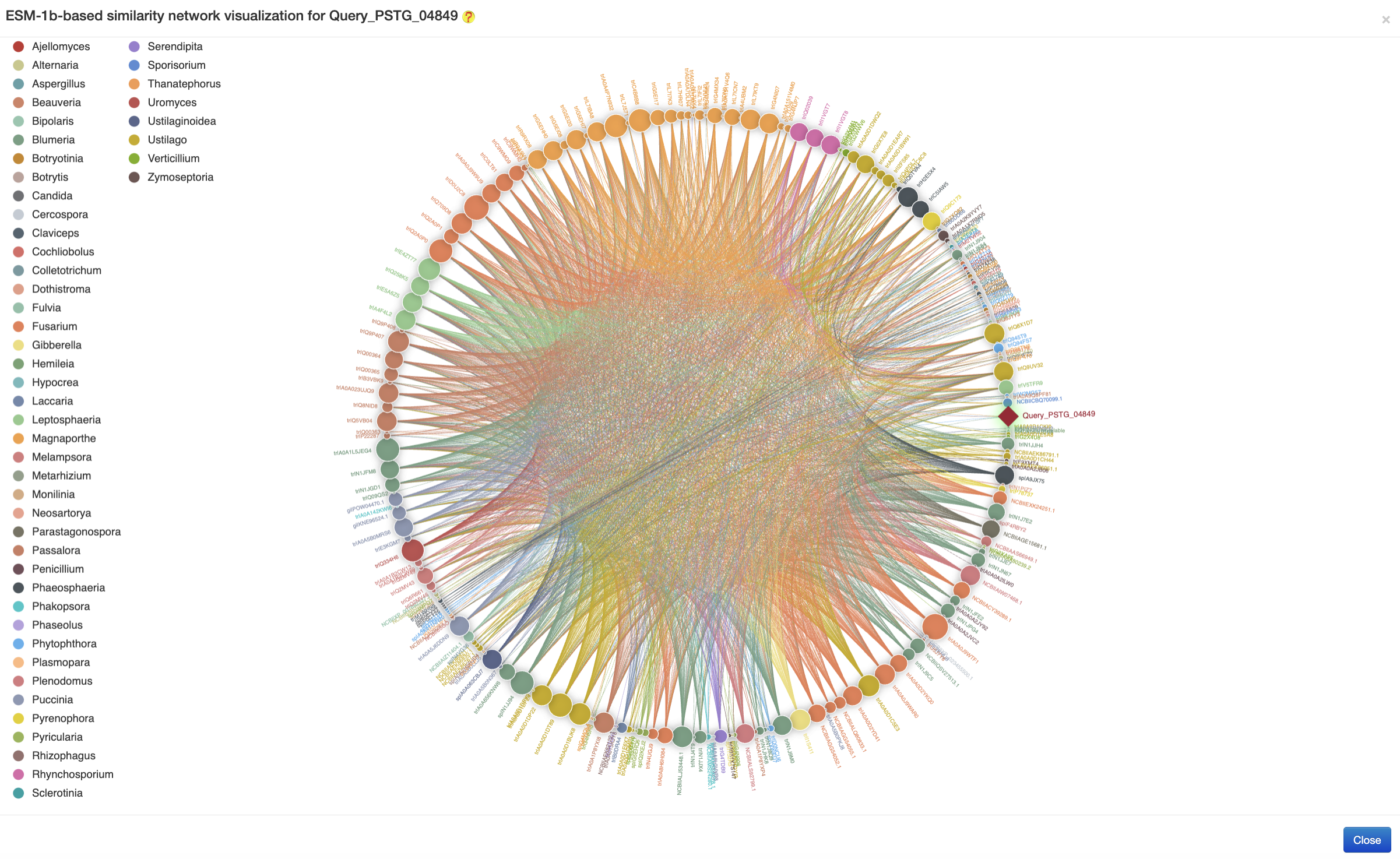
The ESM-1b-based similarity network was generated based on the cosine similarities of ESM-1b features of pairwise fungal effectors (with a threshold of rescaled cosine similarity more than 0.9) to show their high-level connections, followed by the visualization using the ECharts (an example is provided in the following figure)
The relationship analysis was inferred by Bio.Phylo from a Euclidean distance matrix of ESM-1b features of pairwise fungal effectors, followed by the visualization using the phylogram_d3 library (an example is provided in the following figure)